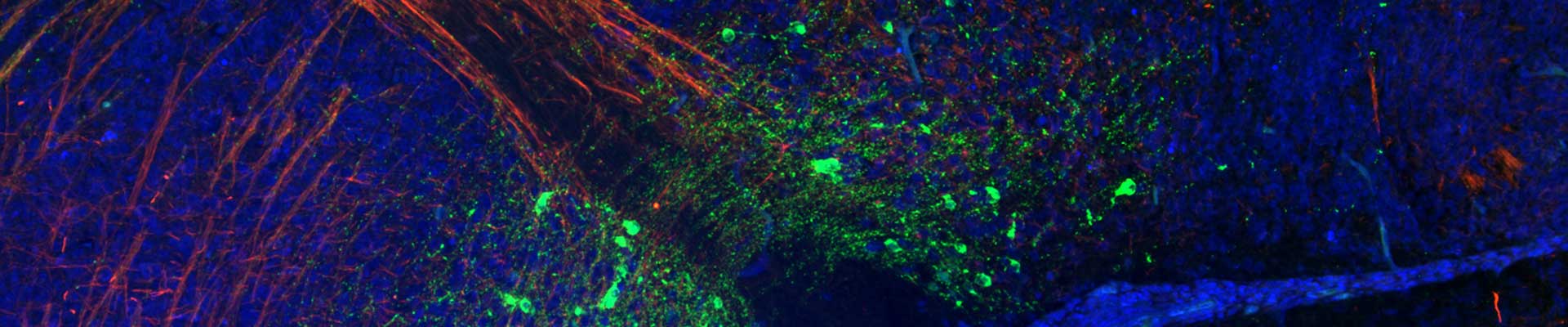
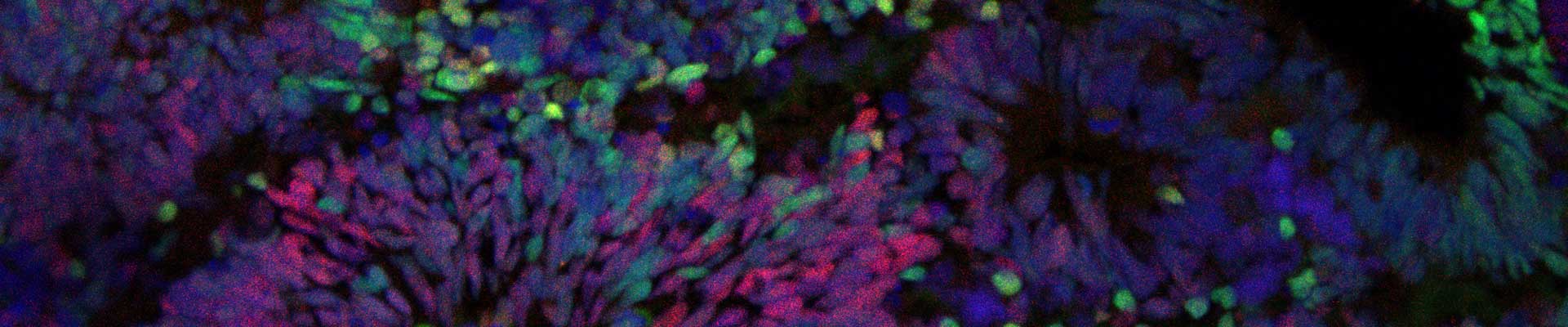
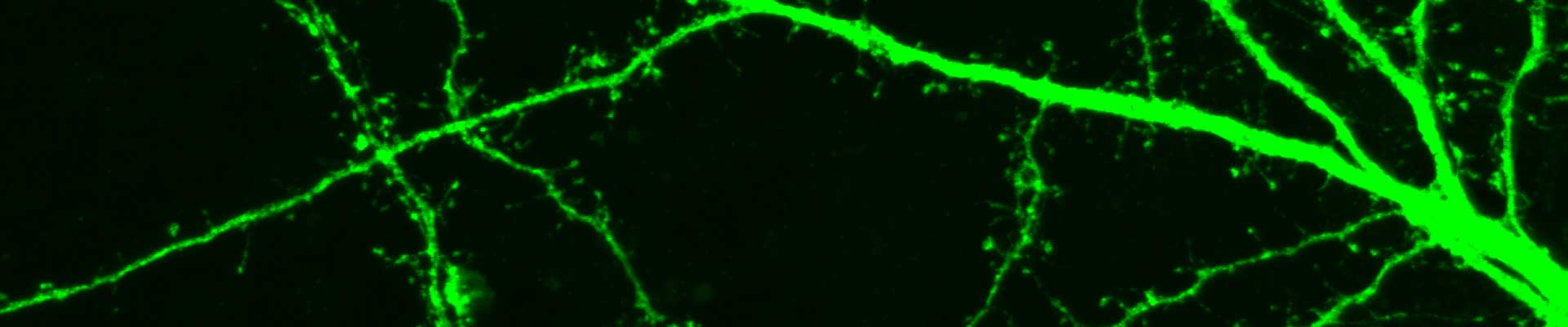
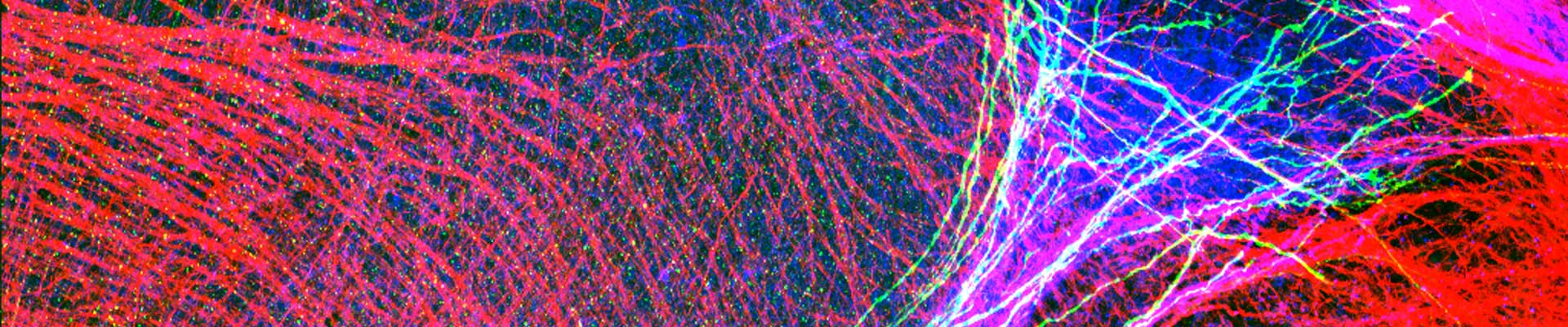
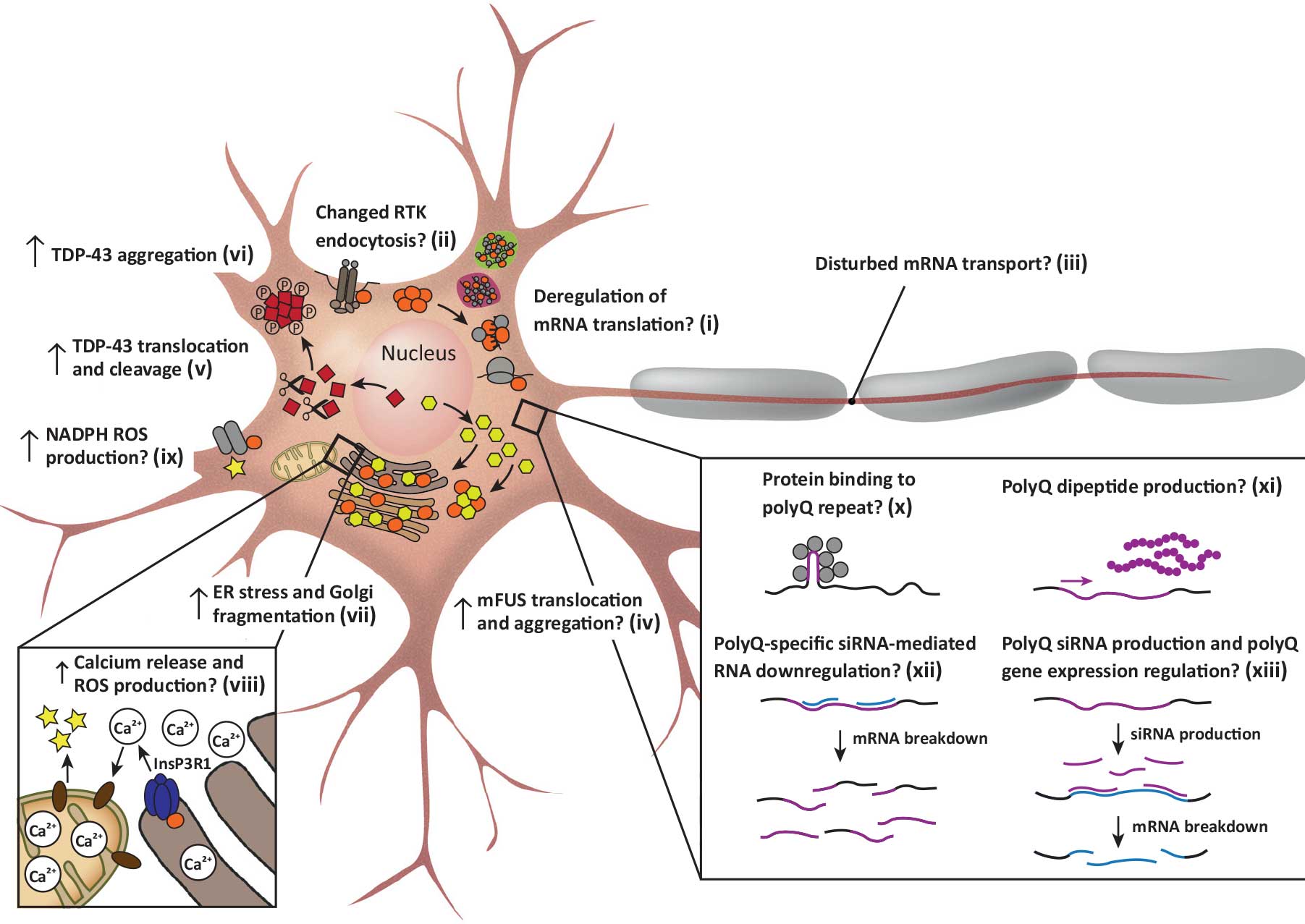
Novel treatment options are needed for this disabling and fatal disease. The lack of treatment in ALS can be attributed to an absence of validated therapeutic targets reflecting an inadequate understanding of disease mechanisms. Elucidating the ALS pathogenic mechanism is therefore essential to pave the road for therapeutic interventions. Several projects in the lab are focused on unravelling the molecular mechanisms defective in in familial and sporadic ALS.
An important starting point of our work are genetic defects detected in ALS patients (e.g. detected in ProjectMinE). Using different molecular cell biological, -omics, and biochemical approaches in different model systems (e.g. IPSC-generated cells (neurons, glia, skeletal muscle) or organoids) we aim to understand how specific genetic defects cause ALS and on basis of this knowledge design therapeutic strategies. This work is in part supported by Stichting ALS Nederland, several E-rare projects and the ALS CURE project. In addition to ALS, we also study other neurodegenerative disorders, including Alzheimer’s and Parkinson’s disease, in part with support from Stichting ParkinsonFonds, Alzheimer Nederland and the JPND project ‘TRIAGE’. We use similar models and approaches to dissect disease mechanisms for these disorders and for designing therapeutic approaches.